Research in the lab is motivated by three broad questions:
- What are the genetic factors that influence human phenotypic variation?
- What environmental and lifestyle factors contribute to this variation?
- How have evolutionary forces shaped this variation and its global distribution?
We attempt to address these questions from an evolutionary perspective by applying computational tools to analyze variation in genomic sequence, DNA methylation, and/or gene expression. These distinct but inter-related sources of information speak to the impact of past evolutionary forces, the biological function of genetic variation, and the effects of an individual’s environment.
Links to all papers and pre-prints from the lab can be found on the Publications page. Below is summary of some of our current major areas of interest.
Epigenomic aging across diverse human populations
Aging is an evolutionary puzzle, and its underlying mechanisms are far from fully understood. However, in just the last few decades, the increased feasibility of assaying epigenetic variation has opened up new avenues to investigate the phenomenon of aging. Unlike genetic variation, which does not change substantially over the course of an individual’s lifetime, DNA methylation patterns change significantly with age. Our research probes DNA methylation variation across populations and species to address classic questions in aging research.
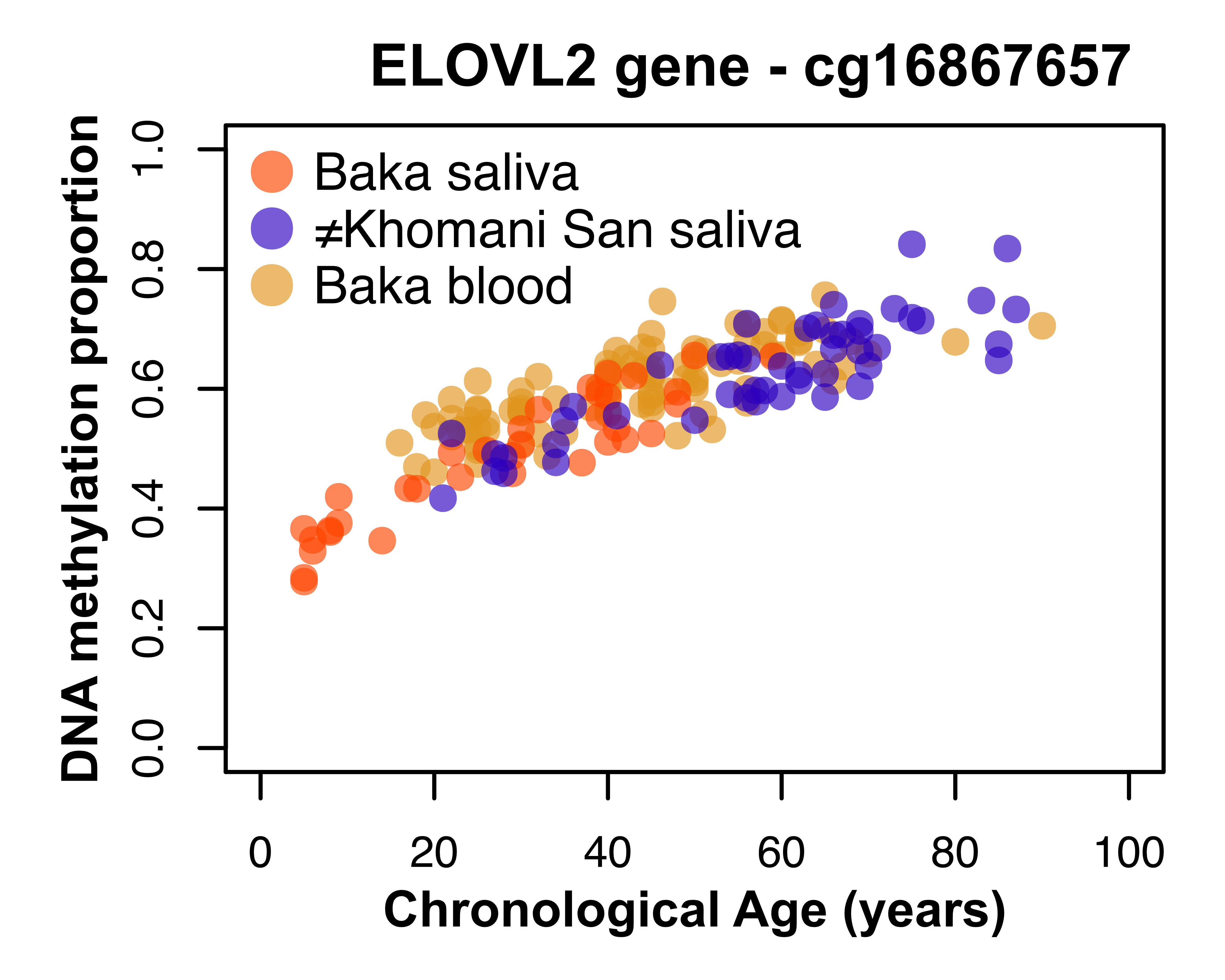
Adapted from Gopalan et al. 2017
In prior work, we examined how DNA methylation patterns change with age, and how these patterns differ across different human populations and across tissue types. We are continuing to investigate the genetic factors that influence epigenetic aging across diverse populations to develop more accurate and universal methods of age prediction. Future research in the lab will leverage DNA methylation data from diverse human populations and from across species to investigate evolutionary constraints on epigenetic aging, how it is impacted by heritable and non-heritable factors, and how it can inform and extend genotype-based methods of trait prediction.
Population genomics of the transition to farming
Throughout history, human populations have been modifying the environment through cultural and technological innovation, thereby shaping their own evolutionary trajectories. An example of this is the adoption of agriculture and/or pastoralism by human societies. These changes coincided with some of the most dramatic demographic shifts in human history – including major population growth, decline, mixture, and movement - while exposing transitioning populations to novel selection pressures. These past events have left distinctive signatures in modern genomes, which we can reconstruct through population genomic analysis.
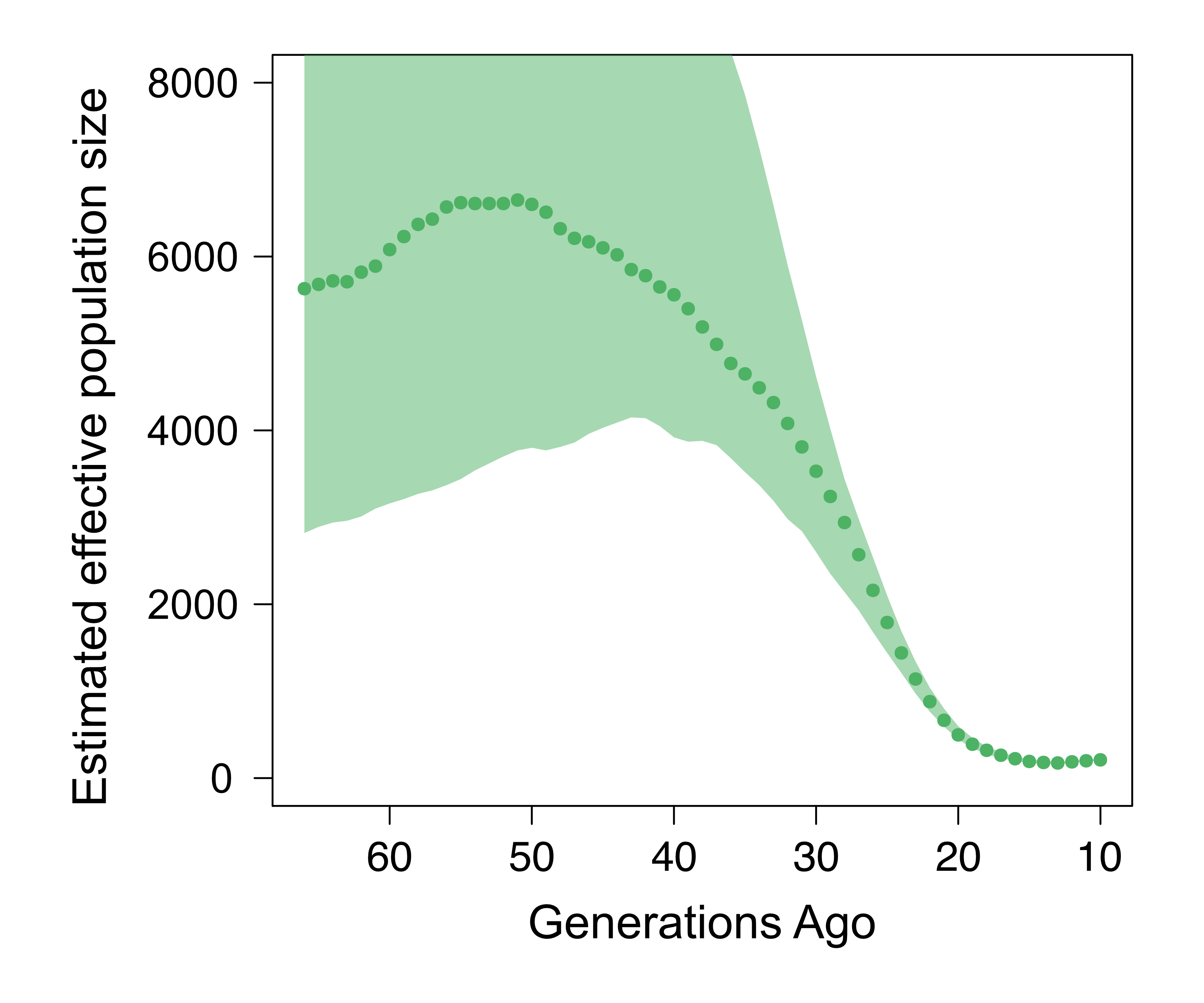
Adapted from Gopalan et al. 2022
Our recent work analyzed population-level genetic data to investigate the history of the Chabu, a population of traditional hunter-gatherers living in the Southwest Ethiopian highlands. We found that the Chabu population has declined significantly over the last few millenia, mirroring trends observed in other Eastern African hunter-gatherers. However, we also observed that this trend was not universal among the descendants of ancient hunter-gatherers of this region. Future work will seek to better understand the demographic history and complex trait architecture of the people of Southwest Ethiopia.
Genomics and epigenomics of domestication
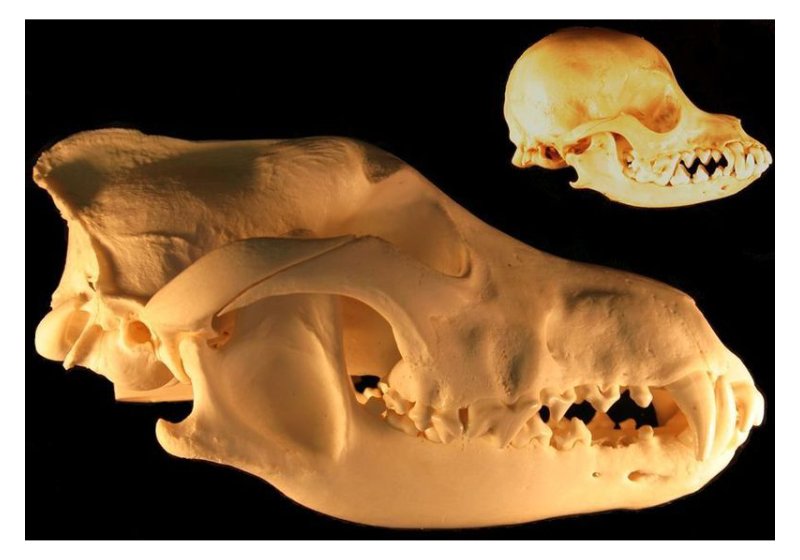
Adapted from Dmccabe, CC BY-SA 3.0, via Wikimedia Commons
The phenomenon of domestication, whereby a wild population becomes dependent on humans over several generations, is frequently associated with strong selection and significant phenotypic divergence over a relatively short period of time. A classic example is the domestication of ancient wolf populations to give rise to dogs, which are genetically, morphologically, and behaviourally distinct from modern wolves. Our research seeks to elucidate the genetic and environmental drivers of these differences by studying epigenetic diversity across canid populations.
Our previous work on ancient dog genomes clarified the timing of domestication and of major demographic events, as well as the evolution of ‘domestication traits’ during the history of dogs. Future work will seek to further clarify the precise ‘tempo and mode’ of the dog domestication process by tracking how the DNA methylation profiles of dogs and wolves diverged over time. By mapping these epigenetic changes to shifts in the environment and/or genetic evolution, we will be able to determine the underlying basis of phenotypic divergence among canid populations.